سارکوم سینوویال
سارکوم سینوویال %۱۰-۵ از کل سارکوم های بافت نرم (STS) را تشکیل می دهد و عمدتا در نوجوانان و جوانان (۱۵-۲۵ سال) رخ می دهد. میزان بروز سارکوم سینوویال ۲،۷۵ در هر ۱۰۰۰۰۰ نفر تخمین زده شده است؛ که شیوع آن در گروه سنی ۲۰ تا ۶۴ سال ۷۴% و در افراد ۶۵ سال و بالاتر ۱۶% است. در جمعیت کودکان، SS شایعترین STS غیر رابدومیوسارکوما است. مردان، کمی بیشتر در معرض خطر قرار دارند.
تظاهرات بالینی
محل درگیری:
سارکوم سینوویال تقریبا در همه جا وجود دارد، اما وقوع درون مفصلی آن بسیار غیر معمول است. این سارکوم می تواند در هر جایی از بافت های نرم به عنوان یک توده در حال گسترش تدریجی، ایجاد شود. اگر چه بسیاری از سارکوم های سینوویال از نزدیک ساختارهای مفصلی منشا می گیرند، نام سارکوم سینوویال یک اشتباه در نامگذاری است؛ زیرا این ضایعات از سینوویوم درون مفصلی منشا نمی گیرند، بلکه از سلول های مزانشیمی اولیه منشا می گیرند؛ با این حال، سارکوم های سینوویال اطراف مفصلی، می توانند به عنوان یک تومور ثانویه به کپسول مفصلی گسترش یابند.
نشانه ها و علائم بالینی:
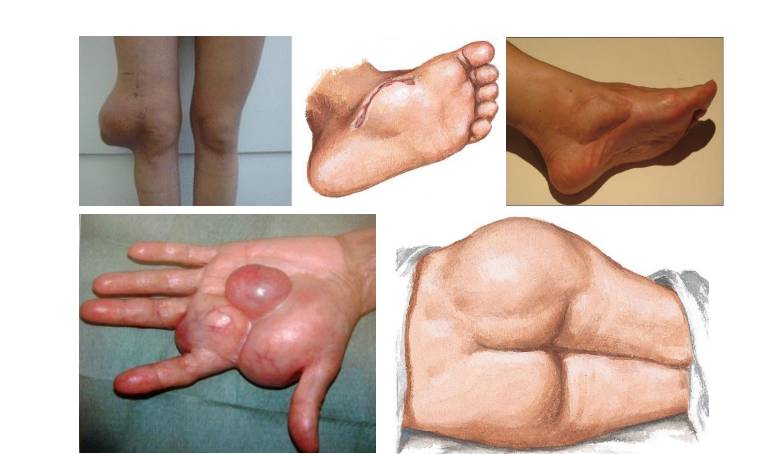
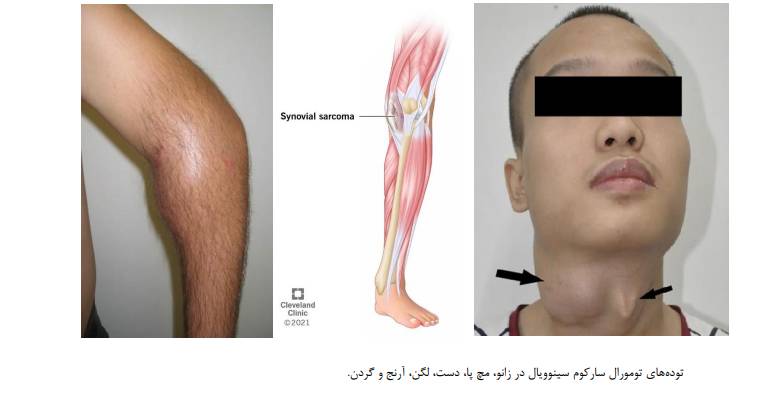
شایعترین علامت بالینی سارکوم سینوویال، توده ای با رشد آهسته در بافت های نرم اندام تحتانی به خصوص در اطراف زانو و مچ پا است. ناحیه سر و گردن، دیواره شکمی، ستون فقرات، مدیاستن، پلورا، ریه ها و سایر اندام ها، مکان های کمتر شایع درگیر هستند. علائم مختلف ممکن است مربوط به محل های مختلف باشد (مانند سختی در بلع و تنفس و یا تغییر صدا در ناحیه سر و گردن).
اگرچه تورم بدون درد شایعترین ظاهر است، درد ناشی از درگیری اعصاب در مراحل پیشرفته ممکن است رخ دهد.
رشد آهسته تومور و بی ضرری ظاهری علائم اغلب منجر به تشخیص با تاخیر می شود و ممکن است در ابتدا با فرآیندهای خوشخیم از جمله میوزیت، سینوزیت، بورسیت یا تاندونیت به اشتباه تشخیص داده شود.
سارکوم سینوویال با تهاجم موضعی و تمایل به متاستاز مشخص می شود. با این وجود، در زمان تشخیص، در کمتر از ۱۰% موارد، متاستاز وجود دارند. با این حال، میزان بالایی از متاستاز دیررس وجود دارد، که در ۵۰% – ۷۰ موارد گزارش شده است. بیشتر متاستازها در ریه ها (%۸۰) ایجاد می شوند، اگرچه استخوان (%۹.۹) و کبد (%۴،۵) مکان های بعدی هستند. در حالی که STS ها در درجه اول توسط مسیر هماتوژن به ریه ها متاستاز می شوند، متاستاز غدد لنفاوی در SS غیرمعمول نیست و بیماری غدد لنفاوی قابل تشخیص بالینی، در ۱ تا %۲۷ از بیماران تازه تشخیص داده شده یافت می شود. مشخص شده که متاستازها در بیماران مسن تر بیشتر است.
تشخیص و تعیین Stage
تصویربرداری:
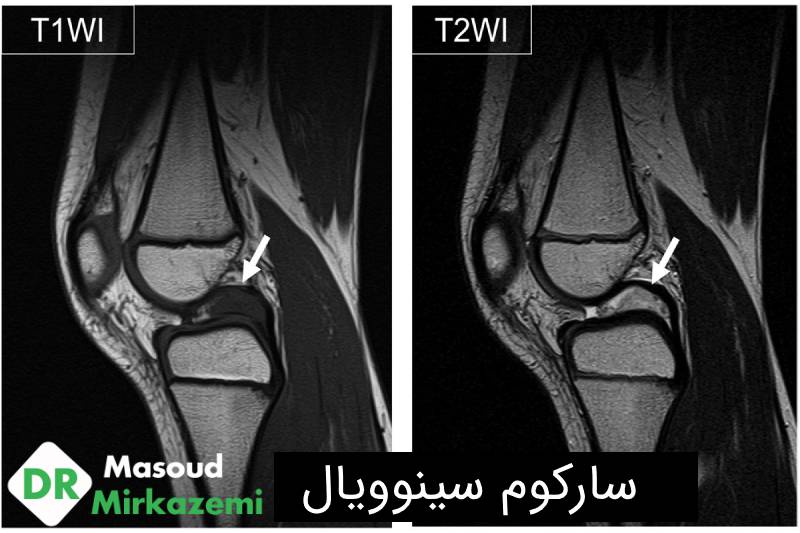
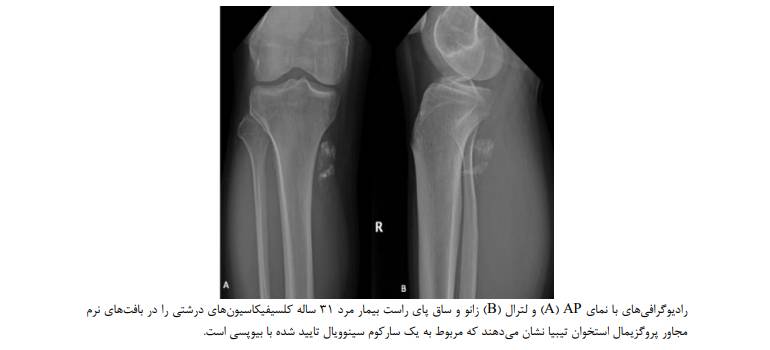
رادیوگرافی های ساده برای تشخیص مورد نیاز نیستند؛ اما معمولا به عنوان بخشی از workup انجام می شوند و می توانند Remodeling مجاور استخوانی، تهاجم استخوانی و کلسیفیکاسیون توده بافت نرم را شناسایی کنند. به طور معمول، سارکوم سینوویال به عنوان یک توده بافت نرم well-defined یا لوبوله در رادیوگرافی های ساده نشان داده می شود. کلسیفیکاسیون های موضعی، به ویژه در اطراف ضایعه، در یک سوم بیماران دیده می شوند. گاهی اوقات، کلسیفیکاسیون گسترده تر ممکن است دیده شود و می تواند تومورهای استخوان ساز (Bone forming) از جمله استئوسارکوم و میوزیت اوسیفیکان (Myositis ossificans) را تقلید کند.
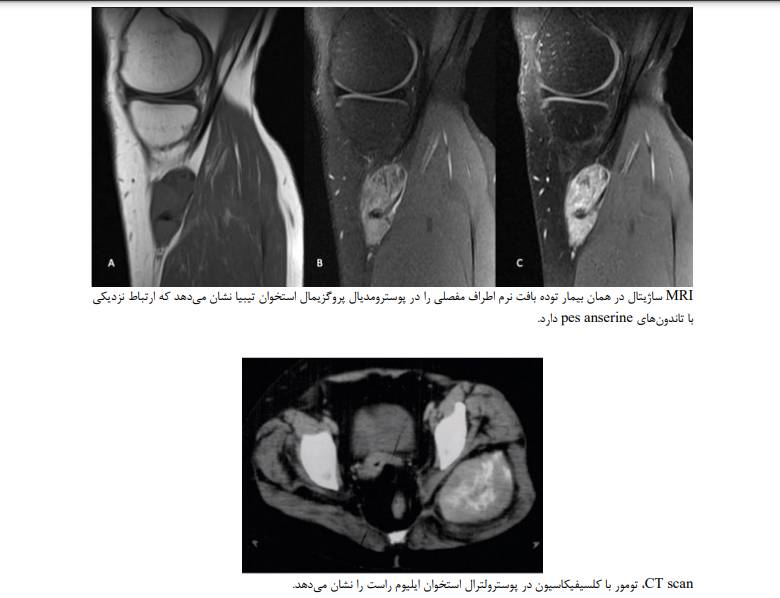
تصویربرداری MRI با و بدون کنتراست گلد استاندارد تصویربرداری تشخیصی سارکوم سینوویال است. MRI محدوده ی محل توده بافت نرم و ادم اطراف آن را مشخص می کند. استفاده از کنتراست گادولینیوم می تواند بین نواحی هموراژیک، نکروتیک و نواحی توموری تمایز ایجاد کند.
اگرچه سارکوم های سینوویال عموما به عنوان یک توده هتروژن غیر اختصاصی وجود دارند، برخی ویژگی هایشان می توانند به تمایز آنها از STS های دیگر کمک کند. سارکوم های سینوویال اغلب به صورت تومورهای جامد well-defined و هتروژن که ماهیت مولتی لوبوله دارند، وجود دارند. شدت سیگنال سه گانه (intensity Triple signal) شامل نواحی hyperintensity ،Isointensity و hypointensity نشان دهنده ی ترکیب نواحی کیستیک و هموراژیک، عناصر سلولی و نواحی فیبروتیک است و می تواند مشخصه ی سارکوم سینوویال باشد. یافته های متعددی در MRI برای پیش بینی ضایعات high grade یافت شده است؛ از جمله عدم وجود کلسیفیکاسیون، وجود خونریزی و شدت سیگنال سه گانه.
در صورت عدم دسترسی به MRI میتوان از CT scan با کنتراست استفاده کرد. CT scan امکان نشان دادن بهتر کلسیفیکاسیون های بافت نرم و واکنش موضعی استخوان را فراهم می کند.
پاتولوژی:
بیوپسی و ارزیابی پاتولوژی برای تمایز سارکوم سینوویال از سایر زیرگروه های STS و تعیین stage تومور مورد نیاز است. مانند تمام STS ها، باید یک بیوپسی قبل از جراحی قطعی انجام شود تا از رزکشن ناکافی و تشخیص اشتباه جلوگیری شود.
با توجه به عوارض کمتر و دقت تشخیصی نسبتا بالای Core needle biopsy، روش ترجیحی بیوپسی، به ویژه برای تومورهای عمیق تر است.
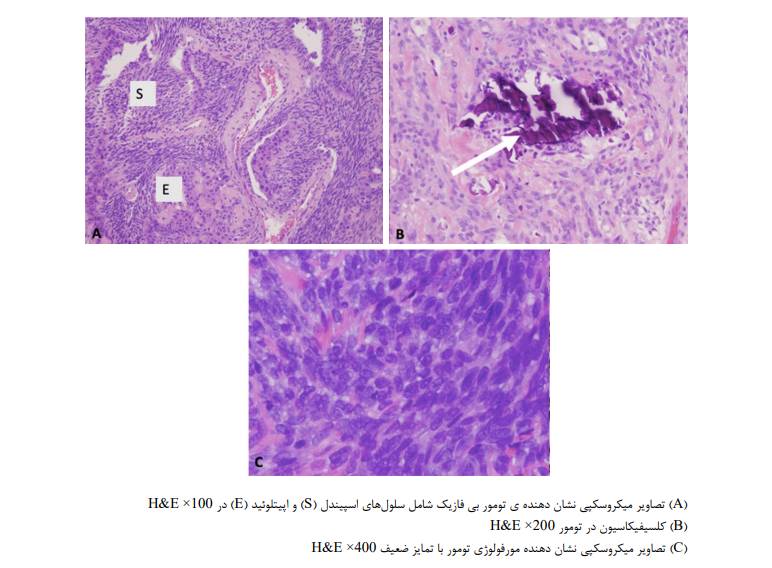
از نظر ماکروسکوپی، SS ها توده های چند وجهی هستند که اندازه آنها بسیار متغیر است. کلسیفیکاسیون، ویژگی های مشترک آنهاست، اما افتراق آنها می تواند دشوار باشد. گاهی اوقات، کیست هایی با دیواره صاف وجود دارند که حاوی مایع مخاطی یا خون هستند. خونریزی و نکروز میتواند در SS با تمایز ضعیف (differentiated poorly) برجسته باشد.
سارکوم های سینوویال از نظر میکروسکوپی در سه زیرگروه بافتی توصیف شده اند: بیفازیک (۲۰ – ۳۰ % ضایعات)؛ مونوفازیک (۵۰ – ۶۰%) به ویژه عناصر سلول اسپیندل مزانشیمی و زیرگروه با تمایز ضعیف (poorly differentiated) ضایعه معمولا well differentiated است و ممکن است توسط یک شبه کپسول (capsule-pseudo) احاطه شده باشد (عمدتا در توده های با ابعاد کوچک)؛ اما همچنین می تواند با حاشیه های poorly defined و پلیلوبوله ظاهر شود و نواحی خونریزی داخل توموری، نکروزه و کیستیک همراه با کلسیفیکاسیون داشته باشد.
پاتولوژی مولکولی:
مشخصه ی سارکوم سینوویال، ترانس لوکیشن پاتوگنومونیک (p11.2 ; q11.2) (x;18) t است که شامل ادغام ژن SS18 (SYT) بر روی کروموزوم ۱۸ به یکی از ژن های سینوویال سارکوما (SSX) بر روی کروموزوم X (معمولا SSX1 یا SSX2) می شود. این مشخصه در بیش از ۹۰% SS ها دیده شده و منجر به تشکیل انکوژن های ادغامی SSX – SS18 می شود.
درمان
همانند تمام سارکوم ها، برنامه درمانی سارکوم سینوویال برای هر بیمار منحصر به فرد است. هر دو متغیر بیمار و تومور در نظر گرفته می شوند تا استراتژی درمانی ایده آل برای هر بیمار تعیین شود.
اساس درمان برای سارکوم سینوویال، جراحی رزکشن با حاشیه های منفی به همراه رادیوتراپی و یا شیمی درمانی، براساس ویژگی های بیمار و تومور است.
جراحی:
در حالی که هیچ دستورالعمل خاصی در مورد حاشیه های منفی ایده آل در سارکومای سینوویال وجود ندارد، مدیریت جراحی مشابه سایر STS است.
برای تومورهای سطحی یا تومورهای کوچک (۵ cm>) که ارتباط نزدیکی با ساختارهای بحرانی ندارند، یک wide resection با حاشیه های منفی (cm 1-2) به تنهایی می تواند کافی باشد. در تومورهایی که ارتباط نزدیکی با ساختارهای عروقی عصبی یا استخوان دارند، اپی نوریوم، ادونتیس یا پریوستئوم به عنوان حاشیه استفاده می شود تا عملکرد اندام حفظ شود. در این موارد، رادیوتراپی برای کاهش خطر عود موضعی ضروری است.
در موارد انتخابی، تکنیک های نجات اندام توصیه نمی شود و ممکن است نیاز به قطع عضو باشد. قطع عضو در بیماران با محل توموری که مستلزم خارج کردن ساختارهای حیاتی ای که منجر به عملکرد ضعیف اندام می شوند است، در نظر گرفته میشود. همچنین بیمارانی که پس از یک excision با آلودگی گستردهای از ساختارهای حیاتی یا مفاصل اصلی مراجعه می کنند، ممکن است نیاز به قطع عضو داشته باشند. در نهایت، بیماران مسنتر یا کسانی که کوموربیدیتی هایی دارند، ممکن است قادر به تحمل یک عمل جراحی عمده نباشند و قطع عضو را میتوان در نظر گرفت.
رادیوتراپی:
پرتودرمانی نئوادجوانت یا ادجوانت برای تومورهای بزرگتر (<5 سانتی متر) یا در هر موردی که ممکن است برای حفظ یک ساختار عروقی یا استخوانی اصلی نیاز به حاشیه نزدیک باشد، توصیه میشود.
پرتودرمانی با شدت تعدیل شده (IMRT) در حال تبدیل شدن به روش ارجح تاباندن پرتو در بیماران مبتال به STS است. ،اجازه میدهد که دوز بالاتری از پرتو به تومور نزدیکتر شود؛ که باعث کاهش حجم پرتودهی به بافت های طبیعی اطراف می شود. نشان داده شده است که استفاده از IMRT پیش از عمل برای STS، عوارض زخم و نیاز به فلپ های بافت نرم ترمیمی را کاهش می دهد. رادیوتراپی ممکن است به صورت مجزا در بیماران با بیماری های متعدد پزشکی یا بیماران مبتلا به بیماری متاستاتیک که در آن خطرات جراحی بیشتر از مزایای بالقوه است، در نظر گرفته شود.
شیمی درمانی و درمان سیستمیک:
برخلاف اکثر STS ها، به نظر می رسد سارکومای سینوویال به شیمی درمانی حساس تر است. به طور کلی، شیمی درمانی برای بیماران با تومورهای پرخطر یا بیماری های پیشرفته در نظر گرفته می شود و تصور می شود در بیماران جوانتر موثرتر باشد. در کودکان و نوجوانان مبتلا به تومورهای متوسط یا پرخطر (مثال <5 سانتی متر، درگیری گره لنفاوی، حاشیههای مثبت)، شیمی درمانی ادجوانت یا نئوادجوانت به طور کلی انجام می شود؛ که شایعترین داروهای مورد استفاده ifosfamide و doxorubicin هستند. داده های اخیر نشان داده است که بیماران کودک یا نوجوان مبتلا به تومورهای کم خطر (درجه ۲ یا درجه ۳< 5 سانتی متر) می توانند با مداخله جراحی بدون درمان سیستماتیک با موفقیت درمان شوند. عوامل جدیدی از جمله مهارکننده های گیرنده ی تیروزین کیناز، تعدیل کننده های اپیژنتیک و ایمونوتراپی ها در کارآزمایی های بالینی مورد بررسی قرار گرفته اند. تاکنون، تنها Pazopanib که یک گیرنده ی مهارکننده تیروزین کیناز است، برای استفاده بالینی تایید شده است. Pazopanib در بیماران مبتلا به بیماری های پیشرفته مورد بررسی قرار گرفته و بهبود بقای بدون پیشرفت را در فاز سوم آزمایش نشان داده است.
پروگنوز
مطالعات فعلی نشان می دهد که نرخ بقای پنج ساله بین ۵۹ تا ۷۵% است. عود موضعی و متاستاتیک سارکوم های بافت نرم عموما در دو سال اول پس از درمان رخ می دهد؛ بنابراین مراقبت و پیگیری در این دوره بسیار مهم است. با این حال، سارکوم سینوویال در این زمینه منحصر به فرد است؛ به این دلیل که خیلی دیرتر عود می کند. مشهود است که عود موضعی به طور میانگین پس از ۳٫۶ سال (بازه ۰٫۵ تا ۱۵ سال) و متاستاز به طور میانگین در ۵٫۷ سال (بازه ۰٫۵ تا ۱۶٫۳ سال) رخ داده است. چندین عامل پیش آگهی کلیدی مستند شده؛ از جمله اندازه، stage و موقعیت آناتومیک تومور، سن بیمار در زمان تشخیص، حاشیه جراحی منفی و رادیوتراپی ادجوانت. تهاجم عروقی یا عصبی، سن بالاتر، اندازه بزرگ تومور و excision برنامه ریزی نشده با پیش آگهی بدتر مرتبط هستند. در یک مطالعه نشان داده شد که اندازه تومور، بقای کلی بدتری را پیش بینی می کند. همچنین نشان داده شده است که موقعیت تومور دارای ارزش پیش بینی کننده است و سارکوم های سینوویال غیرمبتنی بر اندام، بقای کلی بدتری دارند. با این حال، این ممکن است تا حدی به دلیل فقدان علائم اولیه و تظاهرات بعدی باشد. نقش زیرگروه پروتئین انکوژن، متناقض است و به نظر نمی رسد تاثیر مشخصی بر نتایج داشته باشد. طبق داده های اخیر به نظر می رسد که زیرگروه های بافت شناسی از عوامل پیش آگهی هستند، به طوری که زیرگروه بیفازیک بالاترین نرخ بقا پنج ساله و ده ساله را دارد. سارکوم های سینوویال با مناطق با تمایز ضعیف (poorly differentiated)> 20% رفتار تهاجمی تری از خود نشان می دهند.
بهترین نتایج در تومورهایی با ویژگی های بافت شناسی کمتر از ۶ میتوز در میلی متر مربع و بدون نکروز دیده می شوند. اگرچه سارکوم سینوویال در کودکان و بزرگسالان نتایج بالینی مشابهی دارد، اما شواهد فزایندهای وجود دارد که نشان می دهد نتایج در آنها تفاوت دارد و کودکان میزان بقا بسیار بهتری دارند. داده های ثبت شده نشان دادند که نرخ بقای پنج ساله برای کودکان و نوجوانان ۸۳ % است.
منابع:
۱٫ Gazendam, A. M., et al. (2021). “Synovial sarcoma: a clinical review.” 28(3): 1909-1920.
۲٫ Faur, C. I., et al. (2021). “Synovial sarcoma of the extremities: a literature review.”
۱۱(۱۶): ۷۴۰۷٫
۳٫ Fiore, M., et al. (2021). “The biology of synovial sarcoma: state-of-the-art and future
perspectives.” 22(12): 1-22.
۴٫ Caracciolo, J. T., et al. (2019). “Synovial sarcoma of bone: sarcoma typically of soft tissues
presenting as a primary bone tumor.” 14(2): 204-207.
۵٫ Chaparro, E. C., et al. (2018). “Synovial Sarcoma: Imaging findings and prognostic
features”, European Congress of Radiology-ECR 2018.
۶٫ Bakri, A., et al. (2012). “Synovial sarcoma: imaging features of common and uncommon
primary sites, metastatic patterns, and treatment response.” 199(2): W208-W215.
۷٫ Guadagnolo, B. A., et al. (2007). “Long-term outcomes for synovial sarcoma treated with
conservation surgery and radiotherapy.” 69(4): 1173-1180.


دیدگاه خود را ثبت کنید
Want to join the discussion?Feel free to contribute!